

Новость, про которую все пишут исключительно в выражениях «прорыв» и «новая эра в генетической терапии заболеваний». Разумеется, я не могу ее не осветить и, как обычно, немного снижу градус восторга. Потому что, хотя работа [1], о которой идет речь, и действительно вполне себе выдающаяся, до новой эры нам пока что далеко.
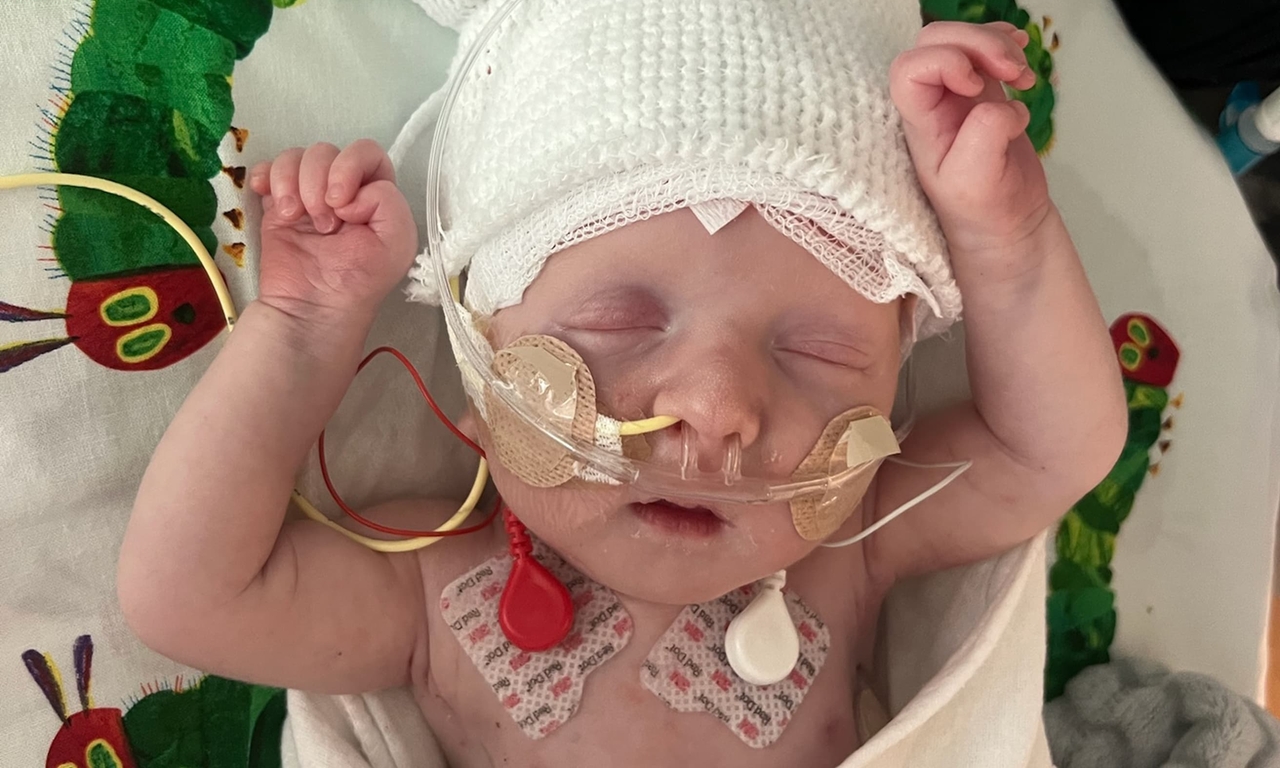
Итак, что произошло. Ученые из Медицинского центра Пенсильваннского университета и детской больницы Филадельфии провели терапию малышу с очень редким наследственным заболеванием при помощи инструмента, который редактирует гены. Я намеренно использовала в предыдущем предложении странное выражение «провели терапию» — для того, чтобы не писать слово «вылечили», так как это была бы неправда.
У малыша, которого родители называют KJ, вскоре после рождения была обнаружена мутация в гене, кодирующем белок карбамоилфосфатсинтетаза 1 (CPS1). Это один из ключевых ферментов так называемого цикла мочевины, или орнитинового цикла, сложной цепочки ферментативных реакций, превращающих опасный аммиак в безвредную мочевину, которая затем выводится с мочой. Аммиак в организме образуется как побочный продукт расщепления белков. Соответственно, у людей с нарушениями орнитинового цикла аммиак накапливается в организме и отравляет его.
В клиническом описании болезни указано, что при легких формах симптомами являются задержка развития и умственная отсталость, а при тяжелых — кома и смерть. Чтобы облегчить проявления болезни, больным пожизненно назначают низкобелковую диету и препараты, выводящие азотистые соединения. Но, несмотря на лечение, многим в конце концов требуется пересадка печени — именно там протекают реакции орнитинового цикла.
Мутация, случившаяся у ребенка по имени KJ, дает одну из самых тяжелых форм болезни — примерно 50% детей с таким диагнозом умирают в младенчестве. Именно поэтому регуляторы разрешили использовать для него генную терапию: в случае KJ потенциальная польза нового лечения перевешивала возможные риски. Терапия, о которой идет речь, нацелена на мутацию, вызывающую болезнь. Она основана на технологии CRISPR/Cas, за которую в 2020 году присудили Нобелевскую премию. Но, в отличие от традиционного варианта CRISPR/Cas, разработчики использовали более мягкую версию, так называемый редактор оснований CRISPR.
Под основаниями имеются в виду азотистые основания, ключевой структурный элемент строительных блоков ДНК — нуклеотидов. Именно тип азотистого основания определяет, какую букву ДНК будут считывать ферменты: А, Т, Г или Ц. И если классический инструмент CRISPR/Cas вырезает из ДНК ошибочные фрагменты, разрезая при этом обе цепи, редактор оснований CRISPR меняет тип азотистого основания при помощи фермента дезаминазы. При этом он надрезает только одну цепь, что многократно снижает вероятность ошибок и проблем при зашивании разрыва.
Благодаря тому, что ученые уже несколько лет работали над созданием платформы для исправления ошибок в геноме на основе редактора оснований CRISPR, после получения одобрения от регуляторов они смогли за полгода сделать модифицированную под мутацию KJ версию инструмента и даже протестировать ее на мышах. Малышу ввели первую дозу в возрасте семи месяцев и вторую спустя еще три недели. Генетические ножницы, как принято называть CRISPR/Cas, попали в печень благодаря тому, что они были упакованы в липидные наночастицы, которые при внутривенном введении склонны накапливаться в клетках печени — гепатоцитах.
В статье в The New England Journal of Medicine (NEJM) описано состояние малыша через семь недель после введения первой дозы. Для точной оценки того, насколько хорошо справился новый инструмент и какой процент клеток печени теперь несут исправленную версию гена CPS1, необходима биопсия, но регулятор счел неэтичным проводить младенцу настолько инвазивную процедуру. Так что ученые и врачи оценивают состояние KJ по косвенным признакам. Малышу вдвое снизили дозу препаратов, выводящих азот, и увеличили содержание белка в диете — и всё это без ухудшения состояния. Кроме того, с момента начала лечения KJ дважды болел простудами: обычно для младенцев с таким диагнозом это смертельно опасно, так как иммунный ответ на вирусную инфекцию активизирует в организме процессы расщепления белков, что при нарушении цикла мочевины может привести к резкому усугублению симптомов.
Но можно ли говорить об окончательном излечении? Нет. Регулятор дал одобрение на введение трех доз препарата, но пока неясно, сколько всего их будет нужно для стабильного улучшения состояния. Кажется, в чем проблема? Вон как хорошо сработали первые две! Но нет. Анализ уже выявил в гепатоцитах KJ одну внецелевую мутацию — то есть редактор оснований CRISPR минимум один раз ошибся. Конкретно эта мутация, по всей видимости, не несет вреда, но со следующими может так не повезти.
Нецелевые мутации — одна из главных проблем всех инструментов генетического редактирования. Каждое новое поколение ошибается меньше, но полностью исключить этот риск нельзя. Кроме того, на сегодня не решена проблема адресной доставки. Липидные наночастицы хорошо накапливаются в печени, но с другими органами и тканями ситуация не такая простая. Пока у ученых нет какого-то универсального решения.
Несмотря на все эти проблемы, генетическое редактирование, разумеется, остается очень многообещающим методом терапии как минимум части генетических заболеваний. Но и надевать розовые очки и переоценивать его возможности тоже не стоит. Чтобы потом не разочаровываться, лучше сначала и не очаровываться.
Ирина Якутенко
1. Kiran Musunuru et al. Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease // The New England Journal of Medicine, May 15, 2025. nejm.org/doi/full/10.1056/NEJMoa2504747
2. The Future of Personalized Medicine is Here: KJ’s Story. chop.edu/centers-programs/genetherapy4inheritedmetabolicdisorders/future-personalized-medicine-here-kjs
